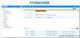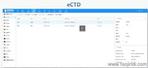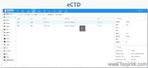
eCTD的技術架構與模塊要求:美國eCTD基于XML技術,嚴格遵循ICH M4框架,分為5個模塊:模塊1(地區行政信息)、模塊2(技術總結)、模塊3-5(質量、非臨床與臨床數據)。其中,模塊1需包含FDA特定的us-regional.xml文件,涵蓋申請編號、聯系人和DMF授權書等行政信息。模塊2-5需與ICH CTD全球統一標準一致,但FDA對文件顆粒度要求更細,例如非臨床研究報告需拆分并標記Study ID。PDF文件需符合FDA v4.1格式規范,包括字體嵌入、書簽層級和超鏈接功能。澳大利亞eCTD注冊咨詢相關技術支持。安徽新藥eCTD服務電話
eCTD生命周期管理與變更提交:歐盟要求eCTD申報資料覆蓋藥品全生命周期,包括提交、補充申請及實質性變更。例如,增成員國需提交“附加成員國序列”,審評時間約52-83天;重大變更(如生產工藝調整)需創建序列并通過CTIS平臺更模塊3和模塊1的GMP證明。技術驗證工具(如EDQM推薦的檢查軟件)需在每次提交前運行,確保XML骨架文件與PDF書簽層級符合規范。此外,電子簽章需符合《歐盟電子簽名法》,并在模塊1中明確標注法律效力。歐洲通用提交門戶(Common European Submission Portal,CESP)是歐盟及成員國藥品監管機構間用于電子化提交申報資料的重要平臺。以下是關于CESP的詳細介紹: CESP是由歐盟藥品監管部門負責人網絡(HMA)合作開發的在線交付系統,旨在為藥品注冊申請者、利益相關方和監管機構之間提供統一、安全的電子提交通道。其設計初衷是簡化跨國申報流程,允許通過單一門戶向多個歐洲國家的藥監部門同時提交申請,避免了重復操作。黑龍江eCTD常用解決方案歐盟NDA注冊申報相關技術支持。
eCTD 4.0版本的過渡與升級:FDA于2023年啟動eCTD 4.0技術試點,2024年9月正式接收申請,計劃2029年完成全過渡。4.0版本改用HL7 RPS標準替代XML,支持雙向通信和跨申請文件復用,例如同一Study ID可在IND和NDA享。模塊1的校驗碼從MD5升級為SHA-256,主干文件由index.xml改為submissionunit.xml,序列號取消前導零(如“1”而非“0001”)。企業需同步更軟件系統以適應架構。DMF與IND申報的特殊要求:針對Type II(原料藥)和Type IV(輔料)DMF,eCTD模塊3需詳細描述生產工藝、穩定性數據,并附分析證書(COA)。FDA要求DMF持有人指定美國境內代理人,確保溝通效率,且LOA(授權書)需明確引用范圍。IND安全性報告(如SUSAR)需通過eCTD模塊5.3.5提交,15天內完成,并嵌入CIOMS或MedWatch表格。2024年指南強調,臨床數據庫需以SAS XPORT格式提交,單個文件超過4GB需拆分并說明規則。
多國審評程序與eCTD遞交途徑的適配:歐盟藥品審評程序包括集中(CP)、分散(DCP)、互認(MRP)和國家程序(NP),eCTD需適配不同程序的遞交要求。例如: 集中審評程序(CP):通過EMA的eSubmission Gateway提交,審評時限約240個工作日,eCTD需包含完整的模塊1-5及多語言標簽文件。 分散審評程序(DCP):需通過CESP(歐盟共同提交門戶)遞交,參考成員國(RMS)主導審評,eCTD需支持多國同步評估的模塊化拆分。 互認程序(MRP):已授權成員國作為RMS,eCTD需包含基線序列(Baseline Sequence 0000)以整合歷史審評數據,并通過CMDh協調分歧。美國NDA注冊申報相關技術支持。

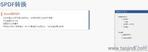
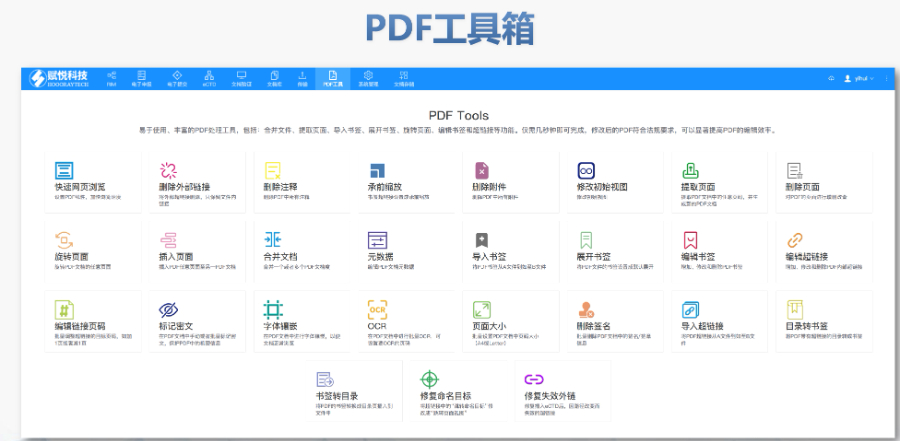
PDF工具箱 批量處理與格式修復 支持PDF合并、拆分、提取頁面、旋轉頁面等操作,可批量修復字體未嵌入、超鏈接錯誤等問題,確保文件符合藥品注冊法規要求。 智能書簽與超鏈接管理 提供書簽導入/導出、超鏈接自動生成(支持關鍵字搜索定位鏈接)、題注超鏈接拖拽式編輯等功能,簡化復雜文檔的導航設計。 文檔轉換與OCR識別 支持Word轉PDF(自動生成書簽、嵌入字體),以及PDF與Word、Excel等格式互轉,集成OCR功能用于掃描件文字識別。 合規性驗證 自動驗證PDF的頁面布局、頁碼連續性、空白頁、目錄層級等屬性,并定位具體錯誤位置,減少人工檢查成本。 安全與協作功能 支持文檔加密、數字簽名、云端同步及多設備共享,滿足企業級文件安全管理需求。澳大利亞eCTD注冊外包相關技術支持。浙江原料藥eCTD服務放心可靠
加拿大IND注冊申報相關技術支持。安徽新藥eCTD服務電話
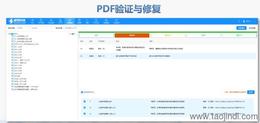
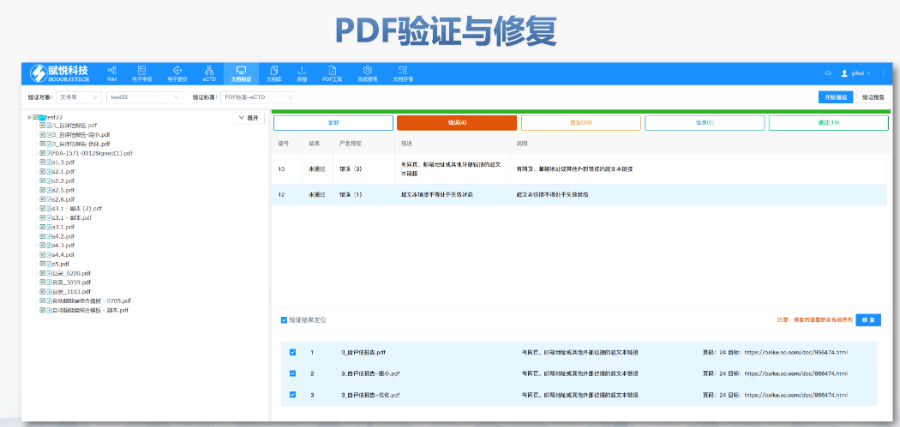
《中國eCTD驗證實踐手冊》作為2025年2月發布的技術指南(發布日期見),為藥品注冊申請人提供了系統化的eCTD申報驗證操作指引。該手冊基于《中國eCTD驗證標準V1.0》的框架,重點覆蓋驗證流程中的六大關鍵領域:基礎識別、文件/文件夾規范、ICH骨架文件完整性、區域性管理信息校驗、研究標簽文件(STF)邏輯性及PDF技術合規性。手冊特別強調對"錯誤警告提示"三級驗證結果的差異化處理策略,指導申請人通過賦悅eCTD軟件進行元數據填報、STF節點配置及擴展節點合規性檢查,同時針對中國特有的注冊類型差異提出模塊化申報資料準備方案。對于PDF文檔,手冊細化到書簽路徑、超鏈接屬性及字體嵌入等技術細節,確保電子資料符合CDE審評系統的解析要求。此外,手冊還結合生物制品與化學藥品的申報差異,明確了3.2.R擴展節點的使用限制,并通過案例解析說明函與申請表生命周期的管理規則。安徽新藥eCTD服務電話